本文介绍了拓扑数据分析(TDA)的基本原理,给出了案例展示,并指出该方法可以高效地进行可视化分析 , 有望为人工智能黑箱提供可解释性 。机器学习和人工智能都是「黑箱」技术——这是使用机器学习、人工智能进行数据研究遭受的批评之一 。虽然它们能自动提供有用的答案,但是却不能给人类提供可解读的输出 。因此 , 我们往往不能了解它们在做什么,又是如何做到的 。
Ayasdi 对提出了解决方法,其中利用了该公司的核心技术——拓扑数据分析(TDA) 。该方法能够提供强有力的、具有详细解释的输出 。然而 , 在这篇文章中,我们将把工作扩展到目前 TDA 的「比较」方法之外 。当前的方法使用的拓扑网络由数据集的数据点(行)构建 。在这项新的工作中,Ayasdi 将特征(列)也融合在网络当中,据此展示了一个改进的、易解释的结果 。
首先介绍一下该解释方法的工作原理 。
假设我们有一个数据集,并且在其中已经辨别出了一些子组 。这些子组可能是数据的一个组成部分(例如,某种疾病有许多不同的形式,比如炎症性肠病 , 或该数据含有一个幸存者/非幸存者的信息),或者说,这些子组是由行集合的某拓扑模型通过分割或热点分析创建的 。
如果选择其中的两个子组,Ayasdi 技术允许研究者根据他们的 Kolmogorov-Smirnov 分数(KS 分数)生成特征列表 。每个特征有两个分布——每个子组各有一个分布 。KS 分数衡量两个子组之间的差异 。与本结构相关的也就是标准统计意义上的 P 值 。
其解释是,排列在第一位的变量是最能区分两个子组的变量,而其余的特征是按其区分能力排列的 。因此,解释机制的输出是一个有序的特征表 。通常,通过查看列表能获得有用的解释,即 , 是何因素导致了不同子组之间的区别 。
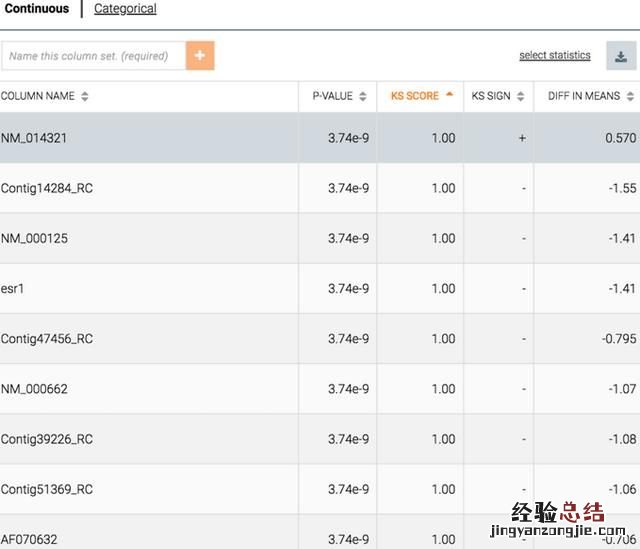
然而,该列表解释起来往往很复杂 。就像 Google 搜索后会得到一长串回复一样 , 人们很可能会发现列表顶部分布不成比例,较低的响应又不为人们所关注 。我们怎样才能进一步提高这些「比较列表」的透明度和可理解性呢?
重要的是,要记?。?Ayasdi 构造的拓扑模型假定给出了一个数据矩阵,以及数据集行的差异性或距离函数 。通常 , 该距离函数是欧几里得距离,但是也可以选择其他距离函数,例如相关距离、各种角度距离等 。获得数据矩阵 M 后,人们可以将它转置为一个新的矩阵 M^T 。其中 , 初始矩阵的列是转置矩阵的行,反之亦然,如下图所示 。
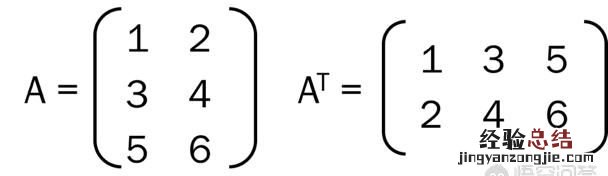
在完成这个操作之后 , 可以为 M^T 矩阵的行集合(即原始矩阵 M 的列)构建拓扑模型 。在集合中,人们可以选择不同的距离函数 。我们不会深入讨论这一点,但总而言之,对任何数据矩阵行的通用可选项对于这个新矩阵也适用 。
现在,假设我们有一个数据矩阵 M , 以及在上述数据集中的一个子组 G 。该子组可能通过先验信息得来 , 也可能通过在 M 矩阵中行的拓扑模型分割得来 。对于矩阵 M 中的每一列 c_i(即转置矩阵 M^T 的每一行),我们现在可以计算子组 G 中每一行的均值,即 c_i 的平均值 。
我们将把它记为 fi,G 。当这个数字包含 i 时,我们在 M^T 的行集合上获得一个函数 。因此,再次重申,M 矩阵中的行的一个子组将在 M^T 的行集合上产生一个函数 。Ayasdi 拓扑模型的功能之一是,通过对应于节点的行,能够利用数据矩阵的行函数的平均值对拓扑模型的节点进行着色 。这对于了解数据属性而言是一个非常有用的方法 。尤其地 , 我们现在可以利用 M^T 矩阵的行集合中子组 G 的着色情况,查看该组的特征 。
请看下例 。
荷兰癌症研究所(NKI)构建了一个数据集,其中包括来自 272 名乳腺癌患者采样的微阵列分析 。本案例中的微阵列分析提供了为研究筛选的一组基因中每个基因的 mRNA 表达水平 。从这些基因中,我们选择了 1500 个表达水平最高的基因 。我们得到一个 272 x 1500 的矩阵,其中 1500 列对应于数据集中具有最大方差的 1500 个基因 , 272 行对应于样本总量 。对于这个数据集,数据矩阵中行集合的拓扑分析已经在 [1] 和 [2] 中进行了 。
我们的拓扑模型展示如下 。
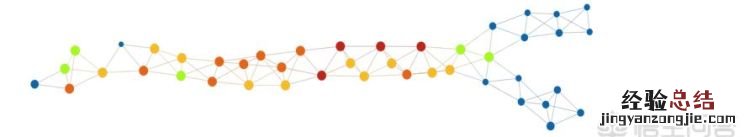
上图表明,拓扑模型包括一个很长的「树干」部分,然后分裂成两个「小枝」 。在数据集中,存在一个名为 eventdeath 的二进制变量 。如果患者在研究期间存活,则 eventdeath = 0;如果患者死亡则 eventdeath = 1 。令人感兴趣的是,患者存活情况与图的结构相对应 。一种方法是通过变量 eventdeath 的平均值进行着色 。其结果如下所示 。
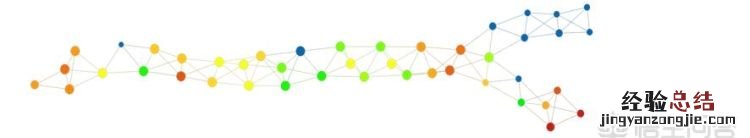
我们可以看到 , 上面的「小枝」呈深蓝色 。这表明 eventdeath 变量值低,实际上其值为零——这意味着每个患者都存活了下来 。然而,下面「小枝」的存活率差得多,尖端节点几乎完全由无法存活的患者组成 。我们希望理解这种现象,看看数据中的哪些特征与「小枝」的产生有关,从而了解变量 eventdeath 的迥异行为 。为此,我们可以从拓扑模型中选择多种不同的子组 。

在上图中 , A 组为高生存率组,B 组为低生存率组,C 组可以表征为与其他两组差异最大的组(根据组间距离进行确定) 。如上所述,基于这三个组,我们可以在 1500 个特征上创建 3 个函数 。
如果我们建立一组特征的拓扑模型,我们可以用每个函数的平均值来给它着色 。下面的三张图片展示了其结果 。
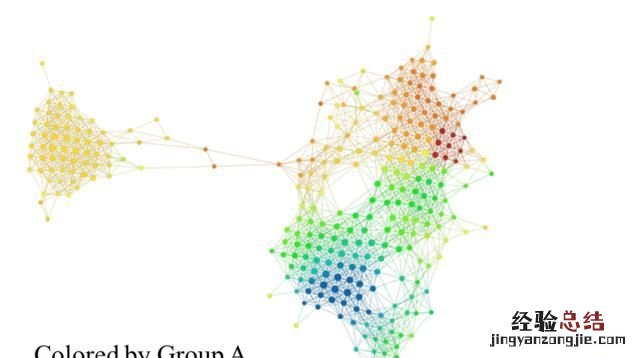


在比较 A 组和 B 组着色情况时 , 我们发现其差异十分显著 。A 组着色后,某个区域呈亮红色 , 而 B 组着色后相应区域呈亮蓝色 。结果可见下图 。左侧的模型是 A 组着色,右侧模型是 B 组着色 。
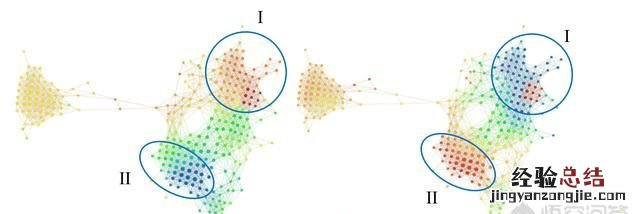
组 I 和组 II 的颜色明显不同 。组 I 在 A 组中主要为红色,而在 B 组中主要为蓝色(小固相区域除外) 。组 II 恰好相反 , 在 A 组中为蓝色,在 B 组中为红色 。这些组可能与高雌激素受体表达有关,其中在组 I 中呈正相关,在组 II 中呈负相关 。众所周知,雌激素受体表达是乳腺癌存活与否的「强信号」 。如果我们比较所有三组(如下图):

我们也可以看到,C 组似乎是 B 组的一个「较弱」形式,其中右上角的蓝色区域面积较?。旅媲虻暮焐先?。在左侧的「岛」上,C 组也显示出比 A 、B 组更强的红色着色 。理解哪些基因参与了 A、B、C 三组右上角的强红色块将是非常有意思的 。此外 , 研究哪些基因参与了左侧「岛」的表达也很有趣 。了解这些基因组需要使用各种基于网络的生物学通路分析的工具 。
总而言之 , 我们已经展示了如何对数据集中的特征空间使用拓扑建模,而不是利用行集合直接从数据集寻找洞察 。具有超过 4 个特征的数据集不能直接使用标准图形技术直观地理解,但是具有成百上千个特征的数据集通过这种方式理解起来却很容易 。该方法能直接识别行为一致的特征组,这通常在基因组和更普遍的生物学数据的分析中存在 。
参考文献
[1] M. Nicolau, A. Levine, and G. Carlsson, Topology based data analysis identifies a subgroup of breast cancers with a unique mutational profile and excellent survival, Proc. Natl. Acad. Sci., vol. 108, no. 17, 7265-7270, (2011).
[2] P. Lum, G. Singh, A. Lehman, T. Ishkhanov, M. Vejdemo-Johansson, M. Alagappan, and G. Carlsson, Extracting insights from the shape of complex data using topology, Scientific Reports 3, Article number 1236, (2013).
依赖大数据和强算力,相当于采用穷举法,就忽视了算法的重要性 。算法是需要循序渐进的研究 。如果方向错了就可能浪费一辈子的时间 。比如说有一根导线环绕地球一圈,某个地方有断点 。如何找到这个断点?根据现在的条件,我们采用穷举的方法,顺序的去找也能找到 。如果采用对分法,很快就能找到 。
【人工智能黑箱问题怎么解决,人工智能算法黑箱问题】
现在所谈论的神经网络,都是标准的相同的结构,他有一个特点就是相邻层的连接 , 是悉数相连的 。因此信息与机构无关 , 只与粘接权重有关,在神经网络中,表现为连接点,递质囊泡的多少 。这种理论暗含着一个潜在的观点,神经网络的结构是由基因遗传的,而不是有后天信息造成的 。那么神经元就没有必要后天产生,实际上大部分神经元是在后天产生的,而且各不相同 。神经干细胞就是产生神经元的重要材料,并非是修补受伤的神经元 。实验证明老年人的大脑,也有新的神经元产生 。这就是大方向 , 错了造成的结果 。






